
A Brief Introduction To Computational Drug Discovery And Its Applications
Namya Mehan — McMaster University Integrated Biomedical Engineering & Health Sciences 2024
Over the past year, we all have been hearing a lot about molecules such as chloroquine, remdesivir, lopinavir/ritonavir, etc. But have you ever wondered what these are, what they aim to do, and where they come from? All of them are small-molecule drugs that were under trial by the World Health Organisation to test their effectiveness against COVID-19. All drugs, small or large molecules, are obtained through a process called drug discovery, by which a drug candidate is identified and partially validated for the treatment of a specific disease.
This process of drug design and discovery involves many steps, such as studying the mechanism of action of various drugs, target selection and validation, lead identification and optimization, drug toxicity, and other mechanical and chemical properties, including pharmacokinetics and pharmacodynamics1.

Given the number of steps in the process of drug discovery, it is evident that it can be extremely time-consuming, risky, and costly. A typical discovery and development process takes about 14 years to introduce a drug to the market, costing about 0.8 to 1 billion USD!1 These statistics exemplify the significant drainage of time and money associated with the cycle of drug discovery and development. But the question is, is there a way to make it any faster and cheaper? Yes.
Like everything else, from calculations to writing, drug discovery can also be made faster by computational methods. Developments in combinatorial chemistry and screening technologies have enabled the screening and synthesis of huge libraries of compounds in a short period of time. Computational drug discovery consists of computer programs and tools for designing compounds, lead identification, and repositories to study drug-target interactions. By using these approaches for various stages of the discovery process, the cost of drug development can be halved! The computational drug discovery approaches that are commonly used can be classified into three categories, structure-based drug design (SBDD), ligand-based drug design (LBDD), and sequence-based drug design.
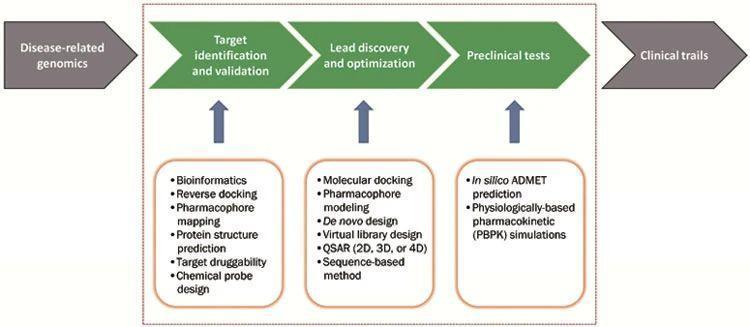
SBDD methods include molecular docking and De novo drug design which both rely on knowledge of the target macromolecule from 3D structures of potential targets. In absence of these 3D structures, LBDD tools provide information about the drug targets and ligand interaction. These tools allow for the construction of predictive models suitable for drug discovery and optimization. Some examples include quantitative structure-activity relationship, pharmacore modeling, molecular field analysis, and 3D similarity assessment. In situations where neither the target nor the ligand information is available, sequence-based approaches that use bioinformatic methods have been developed to identify potential targets and conduct lead discovery. Considering the practical needs of drug discovery, usually, all three approaches mentioned are used in combination to deliver successful results.
There exist several methodologies to support various steps in the drug discovery process. These include using web servers such as TasFisDock that identify drug targets using reverse docking to seek all binding proteins for a given molecule, docking-based virtual screening (SBDD) using GAsDock, a docking methodology with an optimization algorithm that results in more reasonable and robust binding modes between ligands and macromolecules, computational methods such as Cyndi assist in conformational sampling as the algorithms optimize energy and diversity features, virtual libraries play an important role in de novo drug design as they help to overcome challenges in selecting fragment sets for new drug leads. These are some examples of how computational methods are revolutionizing drug discovery.2
While these methods have great potential, the drug discovery process is not completely reliant on computational techniques in a black-box manner. Computational components of research are, and should always be, supplemented and coupled with experimental resources. Future challenges would include the coupling of chemistry and biology with chemoinformatics and bioinformatics, to result in pharmacoinformatics.3 This integration would lead to an increase in the accuracy and effectiveness of computational methods, making them more reliable and trustworthy.
References and Further Reading
- Ou-Yang S-sheng, Lu J-yan, Kong X-Qian, Liang Z-Jie, Luo C, Jiang H. Computational drug discovery. Acta Pharmacologica Sinica. 2012;33(9):1131–40.
- Sliwoski G, Kothiwale S, Meiler J, Lowe EW. Computational Methods in Drug Discovery. Pharmacological Reviews. 2013;66(1):334–95.
- Schaduangrat N, Lampa S, Simeon S, Gleeson MP, Spjuth O, Nantasenamat C. Towards reproducible computational drug discovery. Journal of Cheminformatics. 2020;12(1).

One reply on “How Does Computational Drug Discovery Work?”
Great article!
LikeLike